

















Introduction
China is one of the most populous countries and has the world’s largest medical devices markets. As per a 2018 statistical report, by 2030, China is expected to hold more than 25% share of the global medical device industry at over US$200 billion, which is second to the USA with an expectation of US$300 billion. China’s medical devices market growth rate is driven by a growing customer base in terms of population and health centres, expanding health insurance schemes, the rising prevalence of diseases and disorders, and the rapidly growing geriatric population. Thus, China offers a wealth of opportunities for medical devices companies. However, like all other nations, China has stringent Regulatory requirements and a competitive environment. The medical device manufacturers willing to access the China market must have a clear-cut understanding of the local regulations and technical standards. This article will provide insights on China’s Regulatory approval process for medical devices and the varied premarket or product registration procedures for imported and local domestic products.
Classification of Medical Devices
Depending upon a medical device’s potential risk to patients, the NMPA categorizes medical devices into three (03) classes. As per the degree of risk (from low to high), medical devices are divided into Class I (low-risk), Class II (medium-risk), and Class III (high-risk) in due order. A medical device’s degree of risk is determined comprehensively according to the intended purpose, the pattern of use, structural characteristics, and whether the device is body contacting or not.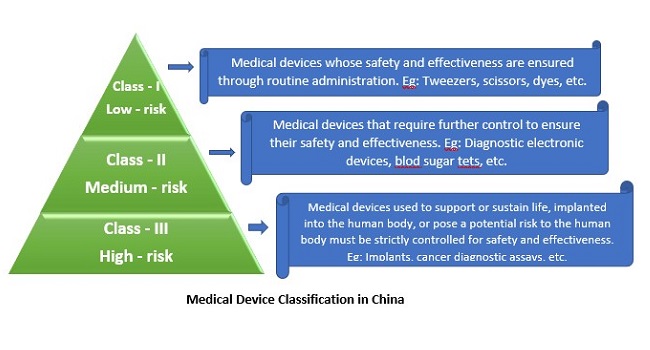
Registration of Medical Devices
- Earlier, China’s medical device registration term was only four (04) years. Currently, it is five (05) years.
- To renew a device’s registration, a renewal application must be submitted six (06) months before the expiration date.
- Overseas medical device manufacturers must provide device samples to the NMPA for testing and meet China testing standard. A test protocol describing the device’s test methodology, parameters, and standards used to prove the device’s safety and performance must be documented. This document is called Product Technical Requirement (PTR).
- Class II and Class III devices require registration. The Class I devices do not require registration but are required to file the device according to the file list. To register Class II and III devices, manufacturers must send the appropriate documents like CE mark, 510(k) letter, ISO 13485 certification, and approved premarket approval application to the NMPA, along with supportive clinical data. All the device information, including labelling and packaging, must be translated into Chinese.
- Foreign manufacturers must hire China-based registration agent/legal agent to register their devices in China. The agent’s responsibilities include:
– providing technical services and maintenance support for the device, if required
– assisting in device recalls, if required
– overseeing the registration and clinical trial process and make sure all the documents are meet China registration requirements
– providing support to the manufacturer if any adverse events occur due to the device Malfunction if required
- Manufacturers must provide the name, address, and contact number of their designated agents in the registration application.
Documentation and Approval
The NMPA requires comprehensive documentation, and all the documents must be submitted in the original language, if applicable and as well as in Chinese. The documents required for new registration and registration extension are mentioned below.
Documents for new registration:
- Regulatory Information include Application form, Product List, associated documents, conformity statement and ect
- Products Description, Applicable scope and contraindications, Product registration history
- Certification documents like EC, EC DoC, and ISO 13485
- Authorization for a legal representative
- The Commitment of an agent and business license of an agent
- List of basic requirements for safety and effectiveness of a medical device
- Clinical evaluation document
- Risk analysis material of products
- Product technical requirement
- Research Information
- NMPA test report
- User manual and label sample
- Conformity declaration and other documents according to the latest regulation or tendency, e.g., cyber security documents
- Quality management system documents etc
Documents for registration extension:
- Application form
- Certification documents like authorization for a legal representative
- The commitment of an agent and business license of an agent
- Declaration on no change of product, copy of original registration license (and change approval license), and attachment
- Conformity declaration
- copies of the original medical device registration certificate and its attachments, and copies of previous medical device change registration (filing) documents and their attachments
- other documents according to the latest regulation or tendency
The new electronic registration system makes it possible to initially register for electronic Regulated Product Submission (eRPS) and be assigned access. While uploading approval documents, manufacturers can use the eRPS to track the documents’ approval status, re-submit the documents or submit answers to the questions raised by the Authority. Electronic submissions can be made for:
- The initial registration/approval of medical devices (including IVD devices)
- Changes to an existing registration of domestic Class III medical devices and foreign Class II and III medical devices
- Applications for clinical investigations for foreign Class III medical devices
- Changes to a medical device’s Instructions for Use (IFU)
- Extension of medical device licenses
- Applications for approval of innovative medical devices
- Changing, correcting, and deregistering licenses for medical devices
Clinical Evaluation
China has stringent clinical trial requirements for medical devices. Clinical evaluation is a process in which the applicant validates whether the devices under registration can meet their intended use and indications based on the information from clinical literature, clinical trials, and clinical experience data. The latest amendment of the regulation have allowed foreign manufacturers to get the clinical trials done in their home country only on the condition to follow all the norms specified in the regulation. Clinical trials are not required for Class I medical devices but are mandatory for Class II and III devices. However, clinical trials may be exempted in one of the following circumstances:
- When the safety and effectiveness of the device can be proved through non-clinical evaluation
- When a medical device’s safety and effectiveness can be demonstrated through the analysis and assessment based on the data obtained from clinical trials or application of a medical device of the same variety
- When the functional mechanism of the device is definite, the production process is well-established, the design is finalized, the marketed medical device of the same variety has been in clinical use for years with no record of serious adverse events, and its conventional purposes of use are not changed
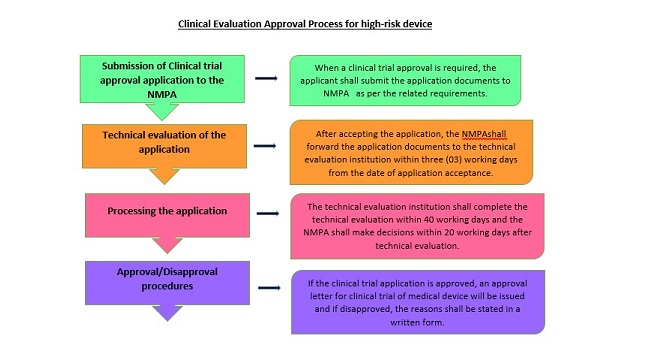
NMPA updates the medical device exemption catalog containing the list of class II and III medical device that are exempted from clinical trials. According to the requirements of the Good Clinical Practice (GCP) for medical devices, the clinical trial of medical devices shall be conducted within a qualified clinical trial institution. The production of samples used for clinical trials shall meet the relevant requirements of a Quality Management System (QMS) for medical devices and shall be filed with the NMPA of the province, autonomous region or municipality directly under the central government where the clinical trial sponsor is located. The local NMPA department that accepts the clinical trial filing shall inform the local NMPA department and the competent health department at the same level of the location of the clinical trial institution of the filing. If the clinical trial of class III medical devices has a high risk to human body, it shall be approved by NMPA under the State Council
Pre-and-Post market Requirements
Instead of focusing on only pre-and-post market requirements, the NMPA emphasizes the administration through the device’s lifecycle. The NMPA is taking effective measures to monitor the device’s performance after being placed on the market, like an adverse event monitoring system, annual periodic risk evaluation report, recalls, and frequent random quality inspections. Therefore, the legal manufacturer is responsible for the safety and effectiveness of the device during its entire lifecycle. The manufacturers are required to establish a QMS and ensure its effective operation and implementation of post-market surveillance and risk control plan, active adverse event monitoring and re-evaluation, establishment and implementation of the product tracing and recalling system.
NMPA’s take on Digital Health
Covid-19 pandemic has fast-forwarded the spread and penetration of digital health technologies with China being in the forefront of this revolution. The adaption of digital health technologies of medical device sector in China is taking place rapidly with considerable changes happening in the regulatory landscape. The government initiatives, advancement of technologies and urge to achieve the “new normal” after the pandemic are driving these swift changes.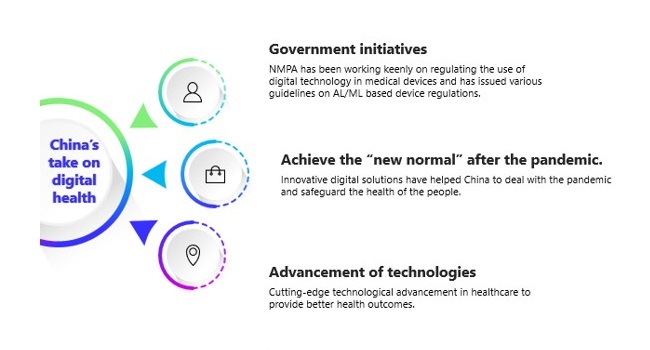
Government initiatives for domestic manufacturers
China’s current contribution in the global device market share is about 20% and is attributed to the aging population, affordability, increased incidence of chronic diseases and lifestyle disorders that offers great opportunities for medical device manufacturers. To strengthen the domestic production of devices, the Chinese government has taken initiatives like setting up medical device industrial zones, rent reductions, settlement bonus or product registration bonus to help domestic manufacturers upscale their business. The Chinese government encourages hospitals to procure domestically produced devices. At present, majority of the domestic manufacturers are small-mid scale businesses manufacturing low value consumables and they dominate the mid-low-end device market. China is still largely dependent on imports for high-end devices. The government’s “Made in China 2025” policy will strengthen the domestic manufacturing of high-end devices in China in coming years.
China device market post Covid 19
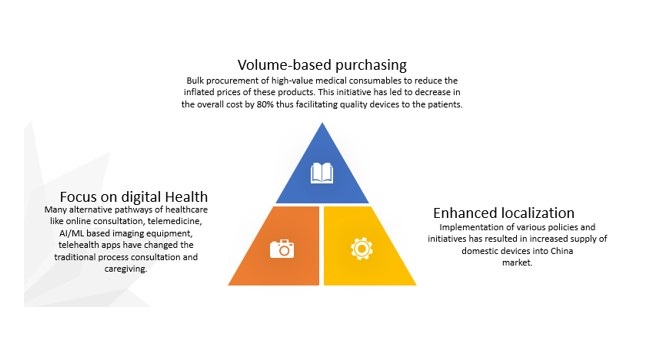
In the view of Covid-19, the Chinese government has taken some steep steps to bring the China medical device industry back to its pace. The government has now enabled three policies for the MedTech market that will ensure its smooth functioning:
China MedTech industry for foreign manufacturers
China is still greatly dependent on foreign manufacturers for high value devices. The foreign manufacturer to enter China market by three possible ways: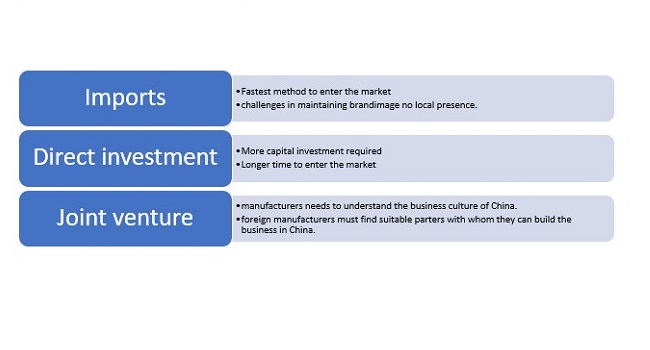
Future trends
With the increasing aging population, lifestyle changes, increased disposable income, China medical device market holds an important place in the global device industry. The stringent regulations ensure that safe and effective devices are available for use. The right regulatory strategy helps the device manufacturers to overcome the challenges and seize the market potential. Adoption of digital health is already in the forefront in China and will disrupt and dominate the global MedTech industry. In the coming years foreign manufacturers will have vast opportunities to enter the Chinese MedTech market owing to the regulatory and governmental reforms. With right entry approach they can capture a stake in this ever-growing market.
Conclusion
China’s medical device market is growing rapidly with accelerating technological transformation in medical devices, overall rising demand for healthcare reforms, increasing aging population, rising prevalence of chronic diseases, and heightened public health awareness. Simultaneously, the NMPA has laid down stringent regulations to ensure safe and effective medical devices are accessible. Therefore, medical device manufacturers willing to enter or expand their presence in China should consider and adopt the industry-specific regulations for a compliant market entry.


{kind=link}